
Business Brief
Literature Review Best Practices Accelerate EU MDR Post Market Surveillance (PMS)Medical device manufacturers continue to work towards achieving compliance with two new guidelines: European Medical Device Regulation (EU) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746, or MDR and IVDR respectively. The MDR went into effect on May 26, 2021, making it critical for device makers who are not yet in compliance to address the new regulatory requirements outlined in this highly detailed and more rigorous regulation. A number of points in the MDR are known to cause issues for manufacturers, including revised rules around literature reviews, clinical evaluation reports, product unique device identifier (UDI) data, and post-market surveillance.
For literature reviews, the MDR calls for manufacturers to demonstrate that they have performed a robust justification to launch a device and keep it on the market.
This is achieved by producing a thorough literature search and retrieving adequate clinical data. The requirements for literature reviews, which are used to support clinical evaluation reports (CERs), are outlined in Annex XIV, Part A of Regulation (EU) 2017/245.
For post-market surveillance, the MDR requires manufacturers to demonstrate that they have a proactive and systematic process in place to collect and utilize product-related information once medical devices are being commercialized. It is important to note that this process is continuous, part of the normal lifecycle of maintaining a device on the market. Guidelines for reporting frequencies are based on device risk class specifications and can be found in the MDR, as laid out in Chapter VII, Articles 83-92 as well as Annex III, Section 1 of Regulation (EU) 2017/245.

Achieving MDR-Compliant Literature Reviews for Medical Devices
The MDR regulation outlines specific requirements for the literature review — a robust process of executing evidence-based research queries that describe medical device product safety, benefit, and risk. Literature reviews are comprehensive searches of published information found in scientific studies related to medical devices on the market. Appropriate search criteria play a critical role in deriving refined and relevant search results for the device in question. Ultimately, the literature review supports CERs and post-market clinical follow-up (PMCF) reports. The CER and PMCF reports are outlined in MDR Annex XIV, Parts A and B, respectively.
Challenges exist for medical device manufacturers performing literature reviews. As mentioned earlier, successful literature reviews depend on the proper framing and subsequent refinement of search criteria. For manufacturers facing compliance requirements for the first time, the literature review introduces new and unfamiliar processes that can change how manufactures procure, collect, and manage information. In addition, new regulatory requirements for literature reviews introduce workload and resource constraints, both of which can impact device readiness project timelines. Literature reviews present a specific obstacle as manufacturers need to demonstrate that they have kept accurate, rigorous, reproducible, and transparent records while continuously monitoring for adverse events and reporting any issues. Finally, the manual literature review is a time-consuming process, with each step in the lifecycle capable of delaying project milestones. More importantly, manual processes can potentially introduce significant errors into regulatory submissions, which may result in costly market delays.
The Medical Device Coordination Group (MDCG) has indicated that MEDDEV 2.7/1 Rev 4 is the key reference document providing guidance and best practices for literature reviews and the literature search protocol. MDCG 2020-13, Section D serves as a complement, providing guidelines on how to conduct the literature review. When possible, manufacturers should consider automating aspects of their literature reviews in order to save time and improve compliance rates.
The use of automated software solutions such as DistillerSR can eliminate many of the manual processes while providing transparent and audit-ready reviews.
Requirements, Challenges, and Best Practices for Post-Market Surveillance (PMS)
Philips Achieves Faster, More Accurate Literature Reviews for CER Submissions with DistillerSR. Read the case study here.
For each medical device that is put on the market, a Post-Market Surveillance (PMS) system must be maintained as part of the quality management system (QMS). The post-market surveillance plan consists of:
- a PMS procedure
- a PMS plan
- a Post-Market Surveillance Report (PMSR) or Periodic Safety Update Report (PSUR), based on the device risk class
The PMS procedure is one or more procedures that medical device manufacturers must create to establish their PMS system. The structure of these framework documents is largely left up to manufacturers but will often take the form of work instructions or standard operating procedures (SOPs). When implementing PMS procedures, manufacturers will likely find a number of additional SOPs internally that will be affected and must be changed. As a result, device makers should consider the broader QMS repercussions of updating their PMS procedures when seeking MDR compliance.
Ultimately, the PMS procedure aims to provide feedback on the benefit-risk determination, clinical evaluation, usability, safety and clinical performance, reportable trends, technical documentation, and post-market surveillance, and when necessary, medical device manufacturers should consider corrective actions for the device in question. Much of this feedback can be sourced through scientific literature.
The PMS plan, outlined in Chapter VII, Article 84, is a comprehensive document that defines the processes and methods involved in collecting and analyzing data related to product performance.
This plan establishes a rigorous, proactive, and systematic process to characterize device performance and compare it to similar medical devices on the market, as well as outlining how data is to be collected. The post-market surveillance plan applies to all device risk classes and should be updated when necessary, though updates can be performed internally and do not need to be made available to notified bodies except upon request for the information.
The PMS plan is part of the MDR technical documentation and outlines the PMS procedure(s) outlined above, as well as the PMSR and the PSUR.
Medical device manufacturers must include in the PMS plan market feedback and customer feedback and complaints, as well as product vigilance, recalls, and literature reviews (including database and register reviews). As a result, the PMS plan will impact multiple other QMS records, including the CER, risk management procedures, corrective and preventive action (CAPA) procedures, trend reporting, PMCF, and instructions for use (IFUs). The MDCG does not provide a solid guidance document or template upon which manufacturers can base their PMS plan. As a result, manufacturers must work to closely follow the MDR guidelines and establish their own planning procedure.
One area of specific confusion among medical device manufacturers unfamiliar with MDR is the revised requirements for PMCF, PMSR, and PSUR.
These documents are distinct from one another and may only apply to specific device risk classifications; however, they do sound similar and can be easily confused with one another.
The PMCF is outlined in Annex XIV, Part B. The PMCF is a specific and proactive form of post-market surveillance required for all Class IIb and Class III medical devices that summarizes clinical evidence from actual and similar devices on the market, including literature publications that highlight product safety and performance. The PMCF consists of both a PMCF plan and a PMCF report. The best practice, in compliance with PMCF requirements, is to follow the guideline documents published by the MDCG, MDCG 2020-07 (PMCF plan) and MDCG 2020-08 (PMCF evaluation report).
Another area of uncertainty for medical device manufacturers relates to the PMSR and the PSUR and their reporting frequency requirements.
The requirements are briefly outlined in Chapter VII, Article 85. This item summarizes results and conclusions of the PMS data, describes corrective actions taken when applicable, and includes both reactive (i.e., complaint-based) as well as proactive (i.e., PMCF) post-market surveillance practices.
The PMSR is only required for Class I devices and becomes part of the technical documentation (TD).

A PMSR is only submitted to competent authorities upon request, rather than being made available for notified bodies during conformity assessment reviews or through EUDAMED. In addition, medical device manufacturers are responsible for updating the PMSR only when necessary.
The PSUR requirements are outlined in Chapter VII, Article 86. This document is similar to the PMSR but applies only to Class IIa, IIb, III, and implantable devices. It also summarizes the results and conclusions of the PMS data and post-market information, vigilance reports, corrective actions, and status of devices on the market.
The PSUR, however, must be updated at least every two years for Class IIa devices and Class IIb non-implantables, and at least every year for Class IIb implantable and Class III devices. In addition, PSURs are submitted during notified body (NB) conformity assessment reviews for Class IIa and Class IIb non-implantable devices, and submitted via EUDAMED for notified body reviews for Class IIb implantables and Class III devices.
It is important to note that while the European Commission does not specify formats for each of these components, there are guidance documents provided by the MDCG that can assist manufacturers with how best to implement these MDR requirements.
Best Practices and Strategies For Compliant EU MDR Literature Reviews
Literature review and post-market surveillance items overlap at two key points during MDR remediation.
The first is in CERs, as outlined in Chapter VI and Annex XIV, Part A. CERs combine key findings in literature reviews and post-market surveillance to summarize clinical evidence and non-clinical data that support device performance and safety.
The second overlap is in PMCF, as outlined in Annex XIV, Part B. PMCFs serve as inputs into CERs and address the post-market surveillance plan by providing evidence of continuous collection of clinical evidence.
PMCFs include data from literature publications on device safety and performance as well as adverse events reports. The literature review can be broken down into a lifecycle, with each step employing unique strategies for ensuring compliance with regulatory requirements:
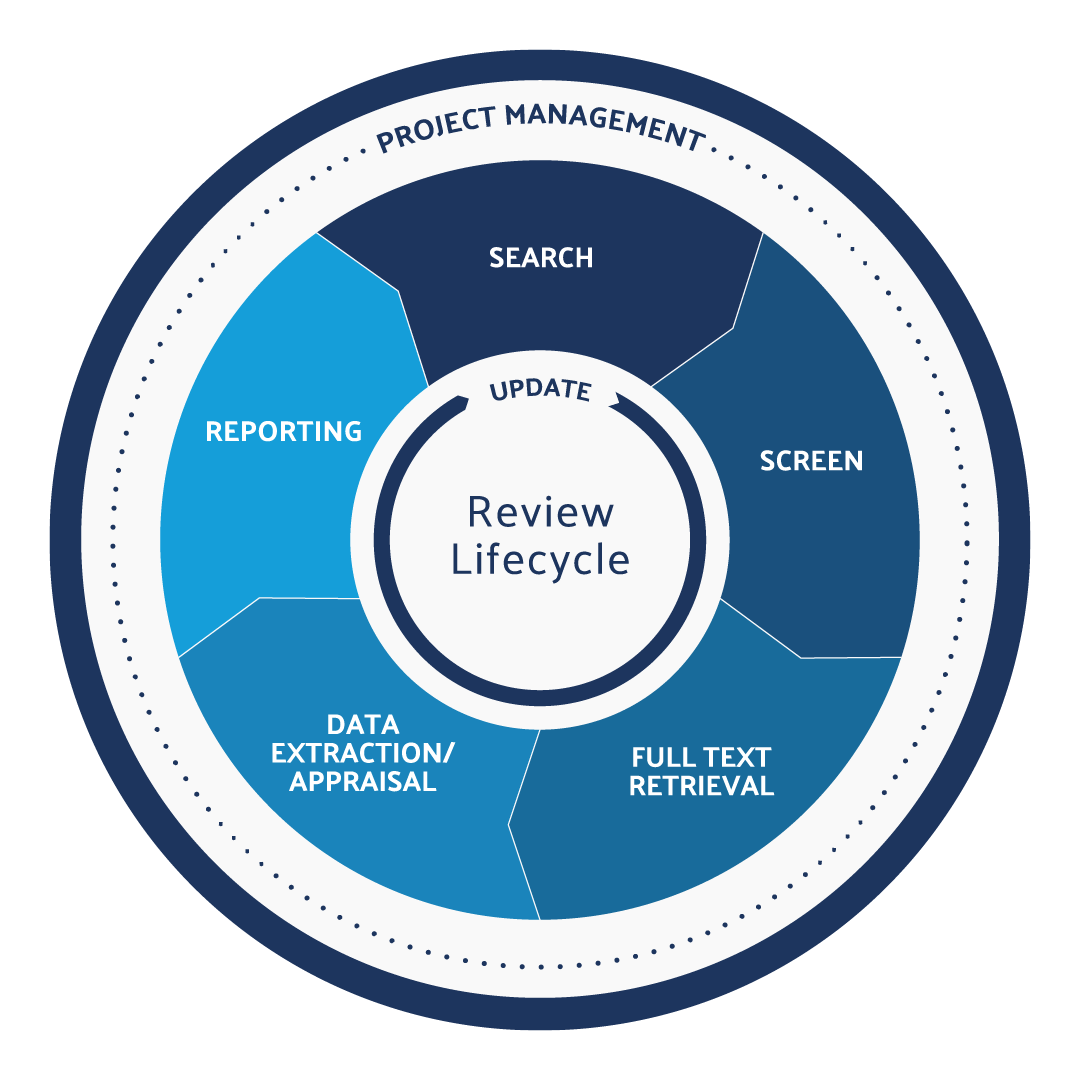
Define the Research Question
Defining an appropriately scoped research question is a critical component of the literature review. Research questions that are too broad may lead to a broad set of search criteria and return too many references that will need to be qualified. Overly narrow research questions may fail to retrieve all relevant evidence- based data and capture the full complexity and function of the medical device product. Furthermore, the research question must yield search results that support the requirements of clinical data as laid out in EU 2017/745 Annex XIV, Part A, Sections 1-4. Specifically, clinical data from the literature review process that supports the CER must include both favorable and unfavorable data and must support the technical, biological, and clinical characteristics of the device.
Defining the research question is the first step in performing a literature summary, but it is also a step that is revisited throughout the literature review lifecycle. MEDDEV 2.7/1 Revision 4, Appendix 5 recommends establishing a literature review protocol that can be used as a key audit tool that will contain the research question. As such, the protocol will define key search terms, databases and sources of data, selection criteria, appraisal and analysis plans, and other components that support the clinical evaluation and performance evaluation reports. Establishing a literature review protocol using this reference as guidance, and including each of these components combined with a well-defined research question, will set medical device manufacturers up to successfully comply with EU MDR regulatory requirements.
Search Relevant Databases
For example, healthcare-focused databases like EMBASE or PubMed should be used, in addition to non-EU-based safety databases such as MAUDE (Manufacturer and User Facility Device Experience). The aim here is to identify the most robust and directly relevant data that supports the safety and efficacy of the medical device product.
Furthermore, medical writers must continuously re-run searches across databases for newly published articles over the course of the review. Using a literature review software platform, such as DistillerSR, that can leverage auto-alerts from data providers to keep the growing set of citations automatically up to date eliminates the tedious task of continuously searching for and uploading newly published or updated records.
Screen References for Relevance
Literature review platforms can facilitate the automatic tracking of inclusion and exclusion decisions of specific data, and some, such as DistillerSR, use artificial intelligence to double-check excluded references and help prevent erroneous decisions.
Initially, the search will generate numerous results that will need to be triaged or prioritized for relevance to the medical device and research question. As such, screening is a resource- intensive part of the review process that can be dramatically reduced through the use of literature review applications. For instance, DistillerSR applies AI to detect and remove duplicate records and to reprioritize unscreened records based on the likelihood of relevance, helping medical writers find relevant references between 40-60% sooner on average than through conventional screening.
Retrieve Full-Text Articles
For example, DistillerSR retrieves all freely available full-text versions of articles automatically, and easily procures those that require purchase through direct integrations with Article Galaxy, RightFind, and e-libraries for the lowest possible cost while ensuring repeat purchases do not occur.
Extract and Appraise the Quality of the Data
Methods for appraising the quality of the data should be registered in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3. Poor data quality can result in one or more of the following scenarios:
- Lack of information on elementary aspects (disclosure omissions)
- Numbers too small for statistical significance
- Improper statistical methods
- Lack of adequate controls
- Improper collection of mortality and serious adverse events data
- Misinterpretation by the authors
- Illegal activities
Standardizing on a comprehensive assessment tool, such as those incorporated in literature review platforms, will ensure all potential data quality shortcomings can clearly be identified and tracked for auditability. DistillerSR comes with a number of industry-standard assessment templates that can be adopted and modified to meet your specific literature review protocol while automatically tracking and associating all decisions and data collected to the appropriate article.
It is important to note that poor data quality does not mean data that shows adverse events. As outlined in EU 2017/745 Annex XIV, Part A, Sections 1-4, the CER must include both favorable and unfavorable clinical data in order to support the technical, biological, and clinical characteristics of the medical device.
Document Purpose and Methods
The purpose and methods should all be documented in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3. A robust, easily traceable literature review protocol will ensure repeatability and transparency in audits, increasing the likelihood of regulatory compliance. In addition, the protocol can serve as a project management guiding document to drive resources supporting the literature review process.
Literature review platforms help to facilitate adherence to specific protocols while automatically tracking each action. DistillerSR, for example, makes it easy to view the provenance of every cell of data and ensures complete transparency and auditability of the entire process, which enables a fully defensible, transparent, and repeatable review.
Monitor for New Literature and Adjust the Research Question, If Necessary
DistillerSR has specific capabilities to ensure you stay up to date with the latest published literature from major data sources (e.g., PubMed, Medline, and EMBASE). It also enables real-time progress monitoring and centrally tracks all project and account activity for a complete audit trail to ensure regulatory compliance.
Learn How Literature Review Automation Improves CER and PER Program Management. Read the business brief here.
Medical Device Industry Trends and Opportunities
Manufacturers must follow the regulatory requirements laid out in the EU-MDR 2017/245, along with industry guidelines provided by MDCG, to make their medical device literature reviews and post-market surveillance programs MDR compliant. Manufacturers that have a larger ecosystem may wish to impose different strategies than smaller companies with fewer products.
One strategy employed by smaller companies simply looking to avoid audit findings and efficiently reach MDR regulatory compliance has been to integrate multiple product lines into the same CER. This must be done strategically to ensure that different devices still meet the MDR requirements for equivalence in technical, clinical, and biological device characteristics, and to ensure auditors can be provided with a rationale for how each product meets standards for the same generic device grouping. The MDR defines a generic device group in Article 2(7) as “a group of devices with the same or similar intended uses or with technological similarities that can be classified even without considering specific features.”
The guidance documents MDCG 2019-13, ISO 13485, and EU Implementation Directive (EU) 2017/2195 provide additional clarity.
Larger companies can also employ these tactics to consolidate larger product families in CERs. Again, establishing technical, clinical, and biological equivalence for each device SKU is critical to justifying these groupings.
Standardization Is Critical to Achieving Compliance with Your Post Market Surveillance Plan
When putting PMCF and PMS requirements into practice, all manufacturers can leverage templates and guidelines that are published by the European Commission and MDCG. For example, ensuring that a compliant PMS plan is in place can be achieved by writing policies that heavily mirror the EU MDR 2017/245. The use of templates can provide a repeatable, consistent, and transparent process to standardize PMCF, PMSR, and PSUR reports across the business. Each can be customized for different product families as needed. Moreover, standardizing literature review processes on a software platform such as DistillerSR further establishes consistency through automated processes, drives internal efficiencies, and ensures all activities are traceable and auditable.
Meeting the regulatory requirements for both literature reviews and post-market surveillance can be challenging for many companies. However, by doing so, manufacturers can ensure that they are well on their way to fully achieving MDR compliance.
Ultimately, leveraging the right tools to do some of the heavy lifting throughout the literature review process and optimizing workflows for continuous data monitoring will be critical for continuous compliance with the EU MDR and, in May 2022, the EU IVDR. Learn more about how do you conduct post-marketing surveillance.
Download the PDF version of this solution brief.
Related Resources

Blog
How DistillerSR has empowered market leaders Philips and Alcon to achieve faster and more accurate literature reviews for CER submissions.

Philips
Philips achieves faster, more accurate literature reviews for CER submissions with DistillerSR.

Blog
Developing a repeatable, transparent process will lead to streamlined, audit-ready, and compliant literature reviews for your PER submissions.